CHE3BPH : Biopharma Assignment Questions and Formula Sheet | Jan L4 Final Exam
| University | La Trobe University (LTU) |
| Subject | Biopharmaceutics |
Question 1 (8 + 9 + (3 + 3 + 3) = 26 marks)
(a) After years of research into stomach cancer, you have finally discovered a new drug that from preliminary studies, appears to be significantly more effective than any of the drugs currently on the market that are used to treat stomach cancer. Briefly outline the stages involved in bringing your new drug candidate to market, indicating what testing requirements are involved in each stage of the process.
(b) What is the difference between pharmaceutical equivalents, pharmaceutical alternatives, and therapeutic equivalents?
(c) (i) What is a biowaiver?
(ii) List THREE types of pharmaceutical formulations that can be considered suitable for a biowaiver?
(iii) Why can a biowaiver be given for these types of formulations?
Question 2 ((1 x 9) + 8 + 14 = 31 marks)
For questions (a) to (i) indicate which statement is correct (each of these questions is worth 1 mark each).
(a) According to Stokes’ law, which of the following changes to a formulation of an oil-in-water emulsion would be expected to decrease the rate of creaming of the emulsion?
- (i) Decrease in the size of the oil droplets.
- (ii) Decrease in the viscosity of the continuous phase.
- (iii) Increase in the difference in density between the oil and water phases.
(b) Which of the following forces leads to attractive interactions between two particles?
- (i) Born forces.
- (ii) Electrostatic forces.
- (iv) van der Waals forces.
- (v) Steric forces.
- (vi) Solvation forces.
(c) When electrolyte is added to a colloidal dispersion:
- (i) The double-layer thickness around the particles is increased.
- (ii) Repulsion between particles is decreased.
- (iv) van der Waals forces between particles are increased.
- (v) The height of the primary maximum is decreased.
(d) Which of the following properties are characteristic of deflocculated suspensions?
- (i) Close packing of the sediment to form a cake.
- (ii) Fast sedimentation rate.
- (iii) Formation of flocs.
- (iv) The solid phase is easily re-suspended.
(e) Stabilisation of oil-in-water emulsions by surfactants:
- (i) Arises because of a reduction of the oil-water interfacial tension.
- (ii) Is a consequence of a decrease of the zeta potential of the oil droplets.
- (iii) Is usually less effective when more than one surfactant is used.
- (iv) Can only be achieved with ionic surfactants.
(f) Rank the following from least surface tension to most surface tension:
- (i) Liquid-liquid (immiscible liquids).
- (ii) Liquid-vapor.
- (iii) Liquid-liquid (miscible liquids).
(g) Which of the following concerning HLB values is NOT true?
- (i) The RHLB of an oil phase of an o/w emulsion is the same as the RHLB for a w/o emulsion using the same oil phase.
- (ii) HLB stands for hydrophile-lipophile balance.
- (iii) A higher HLB value means more hydrophilic.
- (iv) HLB values are assigned to surfactants and oil-like substances.
(h) Oil-soluble surfactants:
- (i) Have high HLB values.
- (ii) Are hydrophilic.
- (iii) Are used as emulsifiers to produce w/o emulsions.
- (iv) Are generally ionic surfactants.
(i) Which one of the following factors is the most important in determining the type of emulsion that forms (i.e. o/w or w/o)?
- (i) The processing and apparatus applied during the production.
- (ii) The HLB value of the emulsifying agents used.
- (iii) The rate of sedimentation of the dispersed drops.
- (iv) The mechanical hardness of the emulsifier film.
(j) From your knowledge of solution thermodynamics, explain why surfactant molecules tend to accumulate at the water-air interface when they are added to water; and at a certain surfactant concentration, referred to as the critical micelle concentration, micelles of surfactant molecules start to form.
(k) Describe the mechanisms by which emulsifiers stabilise an o/w cream formulation. Include in your answer a description of the structure of both the “disperse” and “continuous” phase that forms. Your answer should include a description of both the electrostatic and steric mechanisms by which surfactants stabilize an emulsion. You should also explain why the formation of a stable o/w cream generally requires the use of two surfactants; one with a high HLB and one with a low HLB.
Hire a Professional Essay & Assignment Writer for completing your Academic Assessments
Question 3 (3 + 8 + 3 = 14 marks)
(a) List THREE factors that may affect the rheological properties of an emulsion.
(b) You run a rheogram on an aqueous tragacanth gel. The rheometer is programmed as follows:
- (i) the shear rate is increased from 0 to 400 s-1, at a constant rate, over a period of 60 s;
- (ii) maintained at 400 s-1 for 60 s;
- (iii) then allowed to decrease at a constant rate over a 60 s period until it reaches zero.
The shear stress is plotted as a function of the shear rate. Sketch a diagram of the rheogram you would expect from this gel and provide an explanation for the observed rheological behaviour.
(c) How would you expect the rheogram observed in part (b) to change if the rheometer had been programmed to run under the following conditions:
- (i) the shear rate is increased from 0 to 400 s-1, at a constant rate, over a period of 30 s;
- (ii) maintained at 400 s-1 for 30 s;
- (iii) then allowed to decrease at a constant rate over a 30 s period until it reaches zero.
Question 4 (3 + 3 + 3 + 3 + 3 + 3 = 18 marks)
Zopiclone is a non-benzodiazepine hypnotic agent used to treat insomnia. Consider the following two uncoated tablet formulations for 7.5 mg zopiclone tablets and answer the questions given below. For each question, include a brief explanation to support your answer.
| Ingredient | Tablet 1 | Tablet 2 |
|---|---|---|
| zopiclone | 5 % | 5 % |
| croscarmellose | 5 % | 2 % |
| Lactose | 60 % | 44 % |
| Povidone | 24 % | 45 % |
| stearic acid | 3 % | 3 % |
| Talc | 3 % | 1 % |
(a) Which tablet formulation produced the most friable tablets?
(b) Which tablet formulation produces tablets with the most rapid disintegration rate (i.e. shortest time for complete disintegration)?
(c) Why is a filler required in these tablet formulations?
(d) Which tablet formulation produces the hardest tablets?
(e) As a powder, which tablet formulation flows most freely from the hopper into the die?
(f) As a powder, which tablet formulation is more likely to stick to the tableting equipment during compression?
Question 5 (3 + 2 + 4 + 8 = 17 marks)
Dicloxsig (Aspen) capsules contain 250 mg of dicloxacillin sodium, silicon dioxide, and magnesium stearate. It is used in the treatment of confirmed or suspected staphylococcal and other Gram-positive coccal infections, including skin and skin structure and wound infections, infected burns, cellulitis, osteomyelitis, and pneumonia.
(a) Briefly describe the likely function of each of the excipients used in these capsules.
(b) Why is there no filler used in this formulation?
(c) Would you be able to use this formulation if tablets were produced instead? If not, what excipient(s) might you add to this formulation in order to make it suitable as a tablet formulation?
(d) What factors are important to consider when preparing a homogeneous powder mix? In your answer, describe the phenomenon of demixing and the strategies that can be adopted in order to minimize demixing of powders in the manufacturing process.
Question 6 (8 + 10 = 18 marks)
(a) Apotex-Morphine MR is a sustained release formulation of morphine sulfate and contains the following ingredients: morphine sulfate, lactose, hydroxyethylcellulose, hypromellose, povidone, purified talc, and magnesium stearate.
(i) Identify the likely function(s) of each of the ingredients used and describe how the active is released from the dosage form once it is ingested.
(ii) What are the main advantages and risks associated with using sustained release formulations?
(b) In sustained release dosage forms, multiple unit membrane dosage forms have a number of potential advantages over single unit dosage forms. Briefly outline what these advantages are. Include in your answer a brief discussion on the use of mini tablets compared to other multi-particulates in a multiple unit membrane dosage form?
Question 7 (7 + (6 + 6) + (6 + 6) = 31 marks)
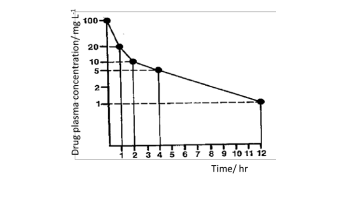
(a) Using the plot of drug plasma concentration (mg L-1) versus time (hr) and knowing that a 500 mg dose was given intravenously, calculate drug clearance using the area method.
(b) A patient is to be started on a continuous infusion of a drug. To achieve an immediate effect, a loading dose is to be administered over 30 minutes and then the continuous infusion is to be begun. From a previous regimen of the same drug, you estimate that the patient’s k = 0.07 hr-1 and Vd = 40 L. Assume that none of this drug has been administered in the last month, so the plasma concentration before therapy is 0 mg/L.
- (i) If the Css(desired) is 15 mg/L, what should the loading dose be?
- (ii) What rate of infusion (K0) should result in a Css of 15 mg/L?
(c) A patient is to be given 100 mg of gentamicin intravenously over 1 hour every 8 hours. If the patient is assumed to have a k of 0.20 hr-1 and a Vd of 15 L:
- (i) How long will it take to reach steady state?
- (ii) For the patient in the question above, what will the peak plasma concentration be after 20 doses?
Buy Custom Answer of This Assessment & Raise Your Grades
Question 8 (6 + 6 + 6 + 3 + 2 + 2 = 25 marks)
Note: Refer to the figure given below in answering questions (a), (b), and (c).
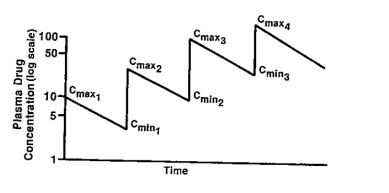
(a) If Cmax1, k, and τ are 100 mg L-1, 0.40 hr-1, and 6 hours, respectively, what is the value of Cmax2?
(b) For the example given in the previous question, what is the value of Cmin2?
(c) What is the maximum concentration after 15 doses if the dose (X0) is 800 mg and the volume of distribution (Vd) is 20 L? Assume that τ is 6 hours and k is 0.50 hr-1.
(d) Using a semi-log plot, sketch the dose-response curve for a drug that would follow a two-compartment model for drug distribution.
(e) On the plot sketched in question (d), identify the location and limits of the ‘distribution’ phase and ‘terminal phase’.
(f) On the plot sketched in question (d), draw the residual line for both the distribution and terminal phase. Using the appropriate terminology, label the y-intercepts and slopes for each residual line.
Formula sheet
1. Diffusion Equations
J = dM/Sdt = D[(C1 – C2)/h]
K = C1/Cd = C2/Cr
dM/dt = DSK[(Cd – Cr)/h]
dM/dt = DSKCd/h = PSCd (sink conditions)
P = DK/h
tL = h2/6D
M = PSCd (t – tL)
dM/dt = (DS/h) (Cs – C)
dC/dt = (D/Vh) (Cs – C)
dM/dt = DSCs /h (sink conditions)
M1/3 = M01/3 – κt
κ = M01/3 2kCs/dρ
C1: solute concentration just inside membrane (donor compartment).
C2: solute concentration just inside membrane (receptor compartment).
Cd: solute concentration (donor compartment).
Cr: solute concentration (receptor compartment).
D: diffusion coefficient. h: membrane thickness.
K: partition coefficient.
M: mass of material passing through membrane. P: permeability.
S: cross-sectional area. t: time. tL: lag time.
V: volume.
2. Pharmacokinetic Equations
(a) One-compartment Model
Cp = Cp0 exp(-kt)
ln Cp = ln Cp0 – kt
Vd = X0/Cp0
CL = k·Vd
t1/2 = 0.693/k
E = (Cin – Cout) /Cin
CLorgan = Q x E = Q x (Cin – Cout)/Cin
Cp: drug plasma conc. Cp0: drug plasma conc. at t = 0. CL: clearance.
E: drug extraction ratio. k (or Kel, k10): elimination rate constant. Vd: volume of distribution.
Q: blood flow. X0: drug dose.
(b) Area Under the Curve (AUC)
AUC = dose administered/drug clearance = X0/CL
AUC = Cp0/k
AUC = X0/Vd·k
Determining the AUC using the trapezoidal rule:
AUC = [{(Cp2 + Cp1)/2} x (t2 – t1)] + [{(Cp3 + Cp2)/2} x (t3 – t2)] + [{(Cp4 + Cp3)/2} x (t4 – t3)] + …….. + Cplast/k
Note: Cplast is the last data point on the Cp versus t graph.
(c) Intravenous Bolus Administration, Multiple Drug Administration, and Steady-State Average Concentrations
Rate of elimination (mg/hr) = CL (L/hr) x Cp (mg/L)
At Cpss: Rate of elimination = Maintenance dose rate (DR) so DR = CL x Cpss
Cpss = F x DR/CL
F = m(drug reached systemic circulation)/m(drug used)
Cpmaxn = Cpmax1(1 – e-nkτ) / (1 – e-kτ)
Cpminn = Cpmax1[(1 – e-nkτ) / (1 – e-kτ)] e-kτ
Accumulation factor = (1 – e-nkτ) / (1 – e-kτ)
Cpmaxss = Cpmax1 / (1 – e-kτ)
Cpminss = [Cpmax1 / (1 – e-kτ)] e-kτ
Cpminss = Cpmaxss e-kτ
Continuous infusion: Cpss = DR/CL
Intermittent dosing: Cpssav = (X0/τ)/CL
Cpssav = X0 / (CL x τ)
X0(new) = (Cpmaxss(new) / Cpmaxss(old)) x X0(old)
Cpmaxss(new) = (X0(new) / X0(old)) x Cpmaxss(old)
Determining the loading dose: X0(loading dose) = Cp(target) x Vd
Cpmaxn: maximum drug plasma conc. (n doses). Cpminn: minimum drug plasma conc. (n doses).
Cpmaxss: maximum steady-state drug plasma conc. Cpssav: average steady-state drug plasma conc.
Cpminss: minimum steady-state drug plasma conc. τ: dosing interval.
(d) Intravenous, Intermittent, and Continuous Infusions
Cp = (K0/CL)(1 – e-kt) (continuous infusion)
K0: rate of drug infusion; t: time since the beginning of the infusion.
Cpss = K0/CL
Loading Dose Equations:
Cp = (X0/Vd)e-kt (IV injection)
Cp = (K0/Vdk)(1 – e-kt’) (continuous infusion), where t’ is the time after beginning the infusion.
Cpmaxss = ((X0/t)/Vdk)(1 – e-kt) (short infusion), where t is the infusion period.
(e) Two-Compartment Model
Vd = VECF + fu(VTBW – VECF)
Vd = VECF + ϕfu(VTBW – VECF)
Cp = Ae-αt + Be-βt
Vd(ss) = V1 + V2
Vc = X0/Cp0 = X0/(A + B)
V(ss) = Vc + (k12/k21)Vc
V(ss) = X0[(A/α2) + (B/β2)]/[(A/α) + (B/β)]2
k21 = (Aβ + Bα)/(A + B)
k = αβ(A + B)/(Aβ + Bα) or k = αβ/k21
k12 = AB(β – α)2/[(A + B) (Aβ + Bα)] or k12 = α + β – k21 – k
Vc(corr.) = Vc(meas.)/[(1 – Hct) + Hct(RBC/P)]
A: “y-intercept” (distribution phase). B: “y-intercept” (elimination phase).
α: 1st order rate constant (distribution phase). β: 1st order rate constant (elimination phase).
fu: unbound fraction of drug in plasma.
k12: rate constant for drug transfer (compartment 1 to 2).
k21: rate constant for drug transfer (compartment 2 to 1).
V1: compartment 1 volume. V2: compartment 2 volume.
Varea (or Vβ): volume of distribution from AUC and β.
Vc: central compartment volume. VECF: extracellular fluid volume.
VTBW: is the total body water volume. V(ss): steady state volume of distribution.
RBC/P: red blood cell/plasma partition ratio.
Hct: hematocrit (volume fraction of red blood cells in blood).
(b)(i) Poorly Water Soluble Drugs and Micronisation
Poorly water-soluble drugs are often micronized in order to improve bioavailability. Micronization increases the surface area of the drug, allowing for better dissolution in the gastrointestinal tract and thus improved absorption.
Potential issues when formulating a micronized active into a capsule or tablet formulation include:
- Possible caking or aggregation of particles.
- Difficulty in achieving consistent dosing due to the fine powder’s flowability.
- Potential for stability issues related to increased surface area.
(b)(ii) Narrow Particle Size Distribution
A narrow particle size distribution is desirable because it ensures uniformity in the dissolution rate, bioavailability, and consistency of the tablet formulation. It also minimizes variations in the drug release profile, leading to more predictable therapeutic outcomes.
(c) Granulation Step in Tablet Manufacturing
Incorporating a granulation step is often necessary in tablet manufacturing because it improves the flow properties of the powder, reduces dusting, ensures uniform distribution of the active pharmaceutical ingredient, and enhances tablet hardness and consistency.
(d) Tablet Thickness Issue
(d)(i) Reasons for Increased Tablet Thickness
Two possible reasons for increased tablet thickness, assuming the formulation composition remains the same, include:
- Increased compression force during tablet formation, which results in thicker tablets.
- Changes in the moisture content or hygroscopicity of excipients, affecting tablet compaction.
(d)(ii) Disintegration Time Issue
If the tablets are passing the active content test but not the disintegration test, it may be due to the following reasons:
- The binder or excipient used in the formulation may have become too strong, causing the tablet to retain its structure and fail to disintegrate.
- Changes in the granulation process, such as insufficient drying or over-drying, leading to changes in tablet porosity, affecting disintegration.
Question 2 (4 + 4 + 5 + 9 + 4 = 26 marks)
(a) Surface Active Molecule
Hexadecyltrimethylammonium bromide is a surface-active molecule due to its amphiphilic nature, which consists of a hydrophobic tail and a hydrophilic head. This structure allows it to reduce the surface tension between liquids and form micelles in aqueous solutions.
(b) Effect of Decreasing Hydrocarbon Portion
If the hydrocarbon portion of hexadecyltrimethylammonium bromide is decreased in length, the micelle size would decrease, and the critical micelle concentration (CMC) would increase, as the molecule becomes less hydrophobic and more prone to aggregation at higher concentrations.
(c) Cloudy Solution at 60°C
If an aqueous solution of a surfactant becomes cloudy at 60°C, it may be due to the surfactant reaching its cloud point, where it undergoes phase separation. This phenomenon is typical of nonionic surfactants.
(d) Emulsifier Mass Estimation
An o/w cream contains 9.0% (w/w) emulsifier and has a required HLB of 11. The surfactants in the cream are sorbitan tristearate (Span 65) and polyoxyethylene sorbitan monopalmitate (Tween 40). If you prepare 200 g of the o/w cream, estimate the mass of each surfactant required to prepare the cream. [HLB (Span 65) = 2.1; HLB (Tween 40) = 15.6.]
(e) Particles in Colloidal Suspension
In a colloidal suspension, particles remain suspended in solution due to their small size, which provides them with a high surface area to volume ratio. This results in the particles undergoing Brownian motion, preventing them from settling. In contrast, particles in a coarse suspension are larger and lack the same stabilizing forces, causing them to eventually settle to the bottom.
Stuck with a lot of homework assignments and feeling stressed ? Take professional academic assistance & Get 100% Plagiarism free papers
Question 3 (4 + (8 + 3) + (6 + 3) + 10 = 34 marks)
(a) Half-lives for Drug Decomposition
To determine the number of half-lives required for 60.0% of a drug to decompose under first-order kinetics, use the equation:
% Remaining = 100 × (1/2)n, where n is the number of half-lives.
(b) Cancer Patient’s Drug Data
(i) Elimination Rate Constant and Volume of Distribution
Using the data provided, calculate the elimination rate constant (k) and the volume of distribution (Vd) for the chemotherapy drug.
(ii) Estimate Clearance
Estimate the clearance of the drug from the body using the data provided and the pharmacokinetic parameters determined in part (a).
(c) IV Drug Dosing
(i) Cpmax after 4 Doses
Calculate the maximum drug plasma concentration (Cpmax) after 4 doses.
(ii) Cpmax after Dose Reduction
Estimate the Cpmax after 4 doses if the dose is reduced to 5.0 mg/kg.
(d) One-Compartment vs. Two-Compartment Model
Describe the differences between a one-compartment and a two-compartment model. Explain the factors that give rise to two-compartment model drug behavior.
Question 4 (6 + 14 + 10 = 30 marks)
(a) Fill Matrices in Soft Gel Capsules
List and describe the three different types of fill matrices used in soft gel capsules.
(b) Modified Release Tablets/Capsules
Discuss the advantages and disadvantages of using modified release tablets or capsules.
(c) Osmotic Pump Modified Release Tablet
Describe how an osmotic pump modified release tablet works and list the main advantages of using this type of modified release dosage form.

- Final Assignment: Migrating FashionOnline’s Infrastructure to AWS: A Strategy for Enhanced Availability and Data Protection
- HRM331: Talent Management – Strategic Shift from the War for Talent to the Wealth of Talent
- Marginalised Populations – The Structural and Cultural Exclusion of People Experiencing Homelessness in Singapore
- CVEN3501 Assignment 2: Groundwater Modelling of Drawdown from a Pumping Bore
- CSCI312 Assignment 2: Conceptual Modelling and Implementation of a Data Warehouse and Hive Queries
- CH2123 Assignment: Fugacity, VLE Modeling & Applications of Henry’s Law
- BAFI1045 Assignment -Constructing and Evaluating Passive and Active Portfolios Based on the Straits Times Index (STI)
- FIN2210E/FIN2212E Group Assignment: Financial Risk Management Analysis of Bursa Malaysia Companies
- FLM101 Assignment: A Film Analysis: Stylistic Techniques and Their Thematic Importance
- HRM Assignment Answer: Talent Transformation in the Age of AI: Turning Challenges into Opportunities via Ecosystem Innovation
UP TO 15 % DISCOUNT